
Статья опубликована в рамках: III Международной научно-практической конференции «Научные достижения биологии, химии, физики» (Россия, г. Новосибирск, 27 декабря 2011 г.)
Наука: Химия
Секция: Биоорганическая химия
Скачать книгу(-и): Сборник статей конференции
- Условия публикаций
- Все статьи конференции
дипломов
ТЕОРЕТИЧЕСКИЕ ПРОСТРАНСТВЕННЫЕ МОДЕЛИ МОЛЕКУЛЯРНОГО КОМПЛЕКСА ПОЛИПЕПТИДА HCGS-2.23 ИЗ АКТИНИИ HETERACTIS CRISPA С ТРИПСИНОМ
Табакмахер Валентин Михайлович
аспирант, ТИБОХ ДВО РАН, г. Владивосток
Гладких Ирина Николаевна
канд. хим. наук, н. с., ТИБОХ ДВО РАН,г. Владивосток
Монастырная Маргарита Михайловна
д-р хим. наук, в .н. с., ТИБОХ ДВО РАН,г. Владивосток
Зелепуга Елена Александровна
канд. физ.-мат. наук, н. с., ТИБОХ ДВО РАН, г. Владивосток
E-mail: zel@pidoc.dvo.ru
Работа выполнена при частичной финансовой поддержке Государственного контракта №16.512.11.2196, гранта РФФИ № 11-04-01179-а, и гранта “Молекулярная и клеточная биология” №09-I-U22-05.
Протеолитические ферменты (протеиназы)присутствуют во всех организмах и составляют от 2 до 4% всех закодированных продуктов гена. Они играют важную роль в различных биологических процессах, таких, как пищеварение, воспаление,свертывание крови, иммунная защита, защита от патогенных инфекций и вирусной репликации - и это лишь некоторые из них. Поскольку протеиназы вызывают необратимое расщепление белков (протеолиз), их активность должна находиться под жестким контролем. Нарушение регуляции протеолитической активности является причиной нарушений в гомеостатическом балансе биологической системы и может привести к непредсказуемому числу негативных биологических последствий. В результате, природой разработан ряд стратегий для управления протеолизом, в том числе посредством активации зимогена, деградации протеаз, а также путем ингибирования протеиназ макромолекулярными ингибиторами. Удивительно, что существует относительно немного принципов конструирования, комбинация которых лежит в основе механизмов действия огромного спектра макромолекулярных ингибиторов протеиназ. Огромные усилия исследователей направлены на изменение специфичности и улучшение эффективности действия ингибиторов, и в значительной степени для этих целей использованы те же принципы дизайна, по которым хорошо работают природные ингибиторы протеиназ.
В данной статье с использованием методов компьютерного моделирования проводится исследование механизма действия полипептида HCGS-2.23 на трипсин. HCGS-2.23 является представителем комбинаторной библиотеки ингибиторов протеиназ семейства Кунитца из ядовитой морской актинии Heteractis сrispa (Тип Кишечнополостные), аминокислотная последовательность которого установлена в нашей лаборатории ранее [8].
Наиболее простым подходом к установлению структурно-функциональных взаимосвязей является сравнение структур новых и уже охарактеризованных соединений. К сожалению, до сих пор пространственная структура ни одного из ингибиторов H. crispa не была установлена экспериментально. Поэтому было решено построить модель пространственной структуры полипептида in silico. Высокая степень идентичности (87%) HCGS-2.23 и ингибитора сериновых протеиназ SHPI-1 из актинии Stichodactylahelianthus, пространственная структура которого установлена методом 1H-ЯМР-спектроскопии (рис. 1, В) [2], позволила использовать для этой цели метод гомологичного моделирования. Теоретическая модель пространственной структуры полипептида HCGS-2.23 была построена при помощи программы SPDBV [7] и веб-сервера SWISS-MODEL [10] с использованием в качестве шаблона пространственной структуры SHPI-1 (PDB ID 1SHP). Для минимизации энергии модели структуры макромолекулы применили программу SPDBV с потенциалом сил GROMOS 96 [13]. Качество модели оценивали с помощью веб-сервера PROCHECK [12]. Суперпозиция полученной модели ингибитора и прототипа SHPI-1 показала, что величина параметров RMSD для 55 Сα-атомов не превышает 0.2 Å. С помощью карты Рамачандрана и сервера PROCHECK полученная модель были проверена на наличие в ней стерических и конформационных затруднений. Было установлено, что 90% аминокислотных остатков имеет благоприятную конформацию, а 10% —допустимую, при этом остатки в неблагоприятной и недопустимой конформациях отсутствовали.
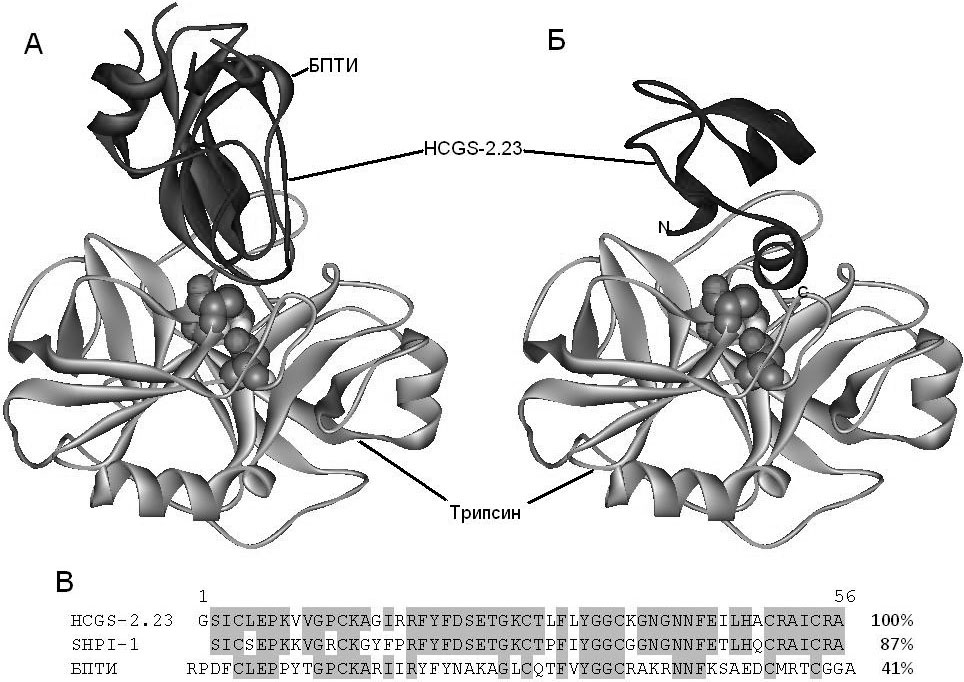
Рисунок 1. Теоретические пространственные модели молекулярного комплекса HCGS-2.23‑трипсин: (А) Суперпозиция теоретической структуры комплекса HCGS-2.23‑трипсин («каноническая» архитектура) с кристаллической структурой комплекса БПТИ‑трипсин (PDB ID 2PTC), (Б) Теоретическая структура комплекса HCGS-2.23‑трипсин (альтернативная архитектура), (В) Выравнивание аминокислотных последовательностей ингибиторов протеиназ: HCGS-2.23 — ингибитор из актинии H. сrispa, SHPI-1 — ингибитор из актинии S. helianthus и БПТИ — ингибитор трипсина из поджелудочной железы Bostaurus. Гомологичные области обозначены серым цветом. Модели молекул представлены в виде ленточных диаграмм. Шаровыми моделями показаны аминокислотные остатки, входящие в активный центр фермента. Визуализация выполнена с помощью программы DS Visualiser 2.5 Accelrys®.
Анализ содержания элементов вторичной структуры в данной модели, проведенный с помощью программы SPDBV, показал, что, подобно структуре наиболее яркого представителя ингибиторов протеиназ семейства Кунитца —БПТИ (бычий панкреатический ингибитор трипсина), она содержит две α-спирали, расположенные на N- и С-концах молекулы, два антипараллельных β-стренда и две большие петли (неупорядоченная структура). Данные о содержании элементов вторичной структуры, полученные с применением компьютерных расчетов, близки данными, установленным экспериментально для ингибиторов протеиназ исследованных ранее in vitro [5, 1]. Таким образом, построенная нами теоретическая модель пространственной структуры полипептида HCGS-2.23 обладает достаточной степенью точности, что позволяет использовать ее для структурно-функциональных исследований.
Теоретические модели комплексов ингибитора с трипсином генерированы с помощью метода молекулярного докинга с использованием ресурсов сервера ClusPro 2.0 [4]. В качестве рецептора использовали структуру трипсинаиз комплекса БПТИ‑трипсин (PDB ID 2PTC). Архитектура большого числа генерированных структурных моделей комплексов HCGS-2.23‑трипсин была аналогична таковой, наблюдаемой в кристаллической структуре комплекса БПТИ‑трипсин. Однако, по результатам расчетов сервера ClusPro 2.0,ингибитор протеиназ HCGS-2.23, кроме комплексов с так называемой «канонической» архитектурой, образует комплексы с альтернативным строением. Так, для комплексов HCGS-2.23‑трипсин наблюдается два типа архитектуры, в которых задействованы либо каноническая связывающая петля, подобно БПТИ (рис. 1, А), либо C‑концевая α‑спираль ингибитора (рис. 1, Б).
Таблица.
Параметры поверхности контактов теоретических комплексов HCGS-2.23‑трипсин с «канонической» и альтернативной архитектурой.
|
Характеристики поверхности контактов (ΔASA) |
«Каноническая» архитектура |
Альтернативная архитектура |
||
|
Трипсин |
HCGS-2.23 |
Трипсин |
HCGS-2.23 |
|
|
Количество сегментов |
11 |
11 |
11 |
11 |
|
Площадь ΔASA (Å2) |
716,61 |
814,43 |
823,99 |
953,91 |
|
% ΔASA |
7,97 |
23,59 |
9,20 |
28,66 |
|
Количество атомов |
136 |
69 |
161 |
75 |
|
Количество а.о. |
27 |
18 |
32 |
20 |
|
% полярных а.о. |
70,37 |
22,22 |
65,62 |
25,00 |
|
% неполярных а.о. |
25,93 |
55,56 |
28,12 |
55,00 |
|
% заряженных а.о. |
3,70 |
22,22 |
6,25 |
20,00 |
|
Планарность (Å) |
6,603 |
2,557 |
6,148 |
2,735 |
|
Водородные связи |
10 |
10 |
7 |
7 |
|
Солевые мостики |
11 |
11 |
6 |
6 |
|
Объемполостей (Å3) |
135,00 |
135,00 |
270,00 |
270,00 |
|
Индекс объема полостей (Å) |
0,09 |
0,08 |
0,16 |
0,14 |
Для валидации полученных нами структурных моделей комплекса был проведен анализ поверхностей контактов (ΔASA). Данные о характеристиках интерфейсов структурных моделей комплекса HCGS-2.23‑трипсиндля «канонического» и альтернативного вариантов архитектуры(см. табл.) соответствуют таковым для экспериментально установленных постоянных белок-белковых комплексов. Кроме того, эти данные свидетельствуют о том, что комплекс с альтернативным строением по большинству характеристик поверхности контактов сопоставим, а по некоторым из них (ΔASA, количество а. о. и атомов на поверхности контактов) превосходит комплекс «канонической» архитектуры.
Как известно, одной из важнейших характеристик белок-белкового взаимодействия является энергия образования комплекса. Программа SEQMOL в 76% случаев позволяет оценить энергию взаимодействия белков, исходя из пространственной структуры их комплекса (экспериментальной или генерированной) с точностью, близкой к экспериментальным данным [9]. Для дискриминации данных структурных моделей комплексов был проведен более глубокий анализ 97 наиболее предпочтительных моделей, полученных после кластеризации результатов геометрического докинга, с целью поиска наиболее энергетически выгодной архитектуры комплекса HCGS-2.23‑трипсин. Для этих моделей с использованием программы SEQMOL были рассчитаны значения изменения свободной энергии Гиббса и константы связывания (Kd) в системе HCGS-2.23‑трипсин. Согласно расчетным данным комплексы с обеими архитектурами, «канонической» и альтернативной (рис. 1, А и Б), соответствуют глобальному минимуму данной системы и имеют близкие значения Kd. Разница в расчетных значениях изменения свободной энергии Гиббса при образовании комплексов с «каноническим» (ΔG = -15,09 ккалл/моль,Kd = 9,78∙10-12 М) и альтернативным (ΔG = -15,19 ккалл/моль, Kd = 8,26∙10-12 М) строением не превышает пределов стандартной погрешности вычислений при использовании экспериментальных методов. Таким образом, полученные результаты компьютерного анализа энергии взаимодействия HCGS-2.23 с трипсином подтверждают наше предположение том, что обе архитектуры комплексов могут быть реализованы и существовать одновременно.
Следует отметить, что интерфейс расчетныхкомплексов HCGS-2.23-трипсин со стороны фермента включает аминокислотные остатки His57, Gly192, Ser195, принимающие участие в каталитическом действии белка. Очевидно, в комплексно-связанном состоянии трипсин не сможет осуществлять катализ гидролитического расщепления полипептидов, что указывает на конкурентный тип ингибирования трипсина полипептидом HCGS-2.23. При этом ингибирующее активность фермента действие HCGS-2.23 должно наблюдаться для обоих вариантов архитектуры комплекса.
В то же время, обобщая результаты анализа поверхностей контактов в «каноническом» комплексе ингибитора HCGS-2.23 с трипсином, проведенного с помощью сервера ProtorP [15], можно сделать вывод, что большой вклад в межмолекулярный интерфейс при связывании HCGS-2.23 с сериновой протеиназой вносят остатки Lys14, Gly16, Ile17. Это коррелирует с данными, полученными экспериментально для БПТИ [11]. Кроме того, расчетными методами установлено, что Arg18 вносит наибольший вклад в связывание полипептидов, образуя четверть ΔASAингибитора и три водородные связи с макромолекулой трипсина. Все это указывает на субстратоподобный механизм действия ингибитора HCGS-2.23 при взаимодействии с классическим сайтом связывания.
В образование комплекса с альтернативным строением со стороны ингибитора наибольший вклад вносят аминокислотные остатки N‑ и C‑концевых α‑спиралей полипептида: Gly1, Ser2, Ile3, His48, Arg51, Ile53, Ala56, при этом наблюдается увеличение поверхности контактов при уменьшении комплиментарности белковых поверхностей в области интерфейса.
Таким образом, методами молекулярного докинга и биоинформационного анализа были получены теоретические пространственные модели комплекса ингибитора HCGS-2.23 комбинаторной библиотеки H. crispa с трипсином. Показано, что HCGS-2.23 образует устойчивые комплексы с Kdпорядка 10-12 М. При этом наблюдается не только «канонический» тип связывания (с участием канонической петли), но и альтернативный (с участием концевых α-спиралей). Показано, что в «каноническом» связывании ингибитора типа Кунитца HCGS-2.23 с трипсином ключевую роль играют Lys14, Gly16, Ile17, Arg18 и имеет место субстратоподобный механизм действия. Установлено, что в рассмотренных структурных моделях комплексов трипсина с полипептидом HCGS-2.23 со стороны сериновой протеиназы задействованы, по крайней мере, 3 аминокислотных остатка активного центра фермента, что говорит о конкурентном типе ингибирования.
Следует отметить, что, несмотря на множество мишеней и разнообразие механизмов ингибирования, большинство ингибиторов протеиназ связывается именно с активным центром протеиназ. По-видимому, данная стратегия обеспечивает высокую эффективность ингибирования, поскольку родственные протеиназы обычно имеют высокую степень гомологии именно в области активного центра и субстратоподобное взаимодействие обеспечивает возможность ингибирования целого ряда протеиназ, тем самым оказывая воздействие на множество биологических процессов. Подтверждением данного феномена, является тот факт, что в среднем один ингибитор протеиназ эффективен для 5 различных протеиназ внутри субкласса ферментов [14]. Тем не менее, не всегда имеет место субстратоподобный механизм ингибирования, как, например, в случае взаимодействия цистатина с цистеиновой протеиназой, когда наибольший вклад во взаимодействие вносят аминокислотные остатки фермента, удаленные от активного центра [3], а также имеет место в случае комплекса HCGS-2.23-трипсин альтернативной архитектуры. Это отличает данный механизм ингибирования от так называемого стандартного субстратоподобного механизма, хотя в данном случае также наблюдается экранирование области активного сайта, но нет непосредственного взаимодействия с каталитической триадой фермента. Такая стратегия, вероятно, обеспечивает еще больше расширение специфичности действия ингибитора.
Как известно, многие ингибиторы протеиназ кровососущих паразитов кроме основного сайта связывания имеют вторичный сайт связывания (экзосайт) за пределами активного сайта, который также важен для ингибирования [16]. Данная стратегия имеет несколько преимуществ. Во-первых, она обеспечивает увеличение поверхности контактов белок-белкового взаимодействия, что позволяет увеличить аффинность и, во-вторых, расширяет спектр возможных мишеней, воздействуя на специфичность ингибитора [6]. Вероятно, подобный механизм действия реализуется и в случае исследуемого нами полипептида HCGS-2.23 —одного из представителей комбинаторной библиотеки ингибиторов протеиназ типа Кунитца актинииH. crispa. По-видимому, в процессе эволюции полипептид HCGS-2.23, обеспечивая защиту организма-продуцента от широкого разнообразия протеиназ жертв и хищников, использовал преимущества наличия экзосайтов не только для максимального расширения спектра специфичности, но и для сохранения достаточно высокой аффинности, о чем свидетельствуют сравнимые значения изменения свободной энергии Гиббса при образовании комплексов трипсина с классическим и альтернативным сайтами связывания ингибитора. Наличие двух сайтов связывания, взаимодействующих с областью активного центра фермента, но использующих при этом различные механизмы действия, позволяет предполагать, что полипептид HCGS-2.23 может ингибировать активность не только трипсиноподобных протеиназ, но других протеолитических ферментов. Это, безусловно, представляет большой теоретический интерес и требует дальнейших экспериментальных исследований.
Список литературы:
1.Вакорина Т. И., Гладких И. Н., Монастырная М. М., Козловская Э. П. Конформационная стабильность ингибитора сериновых протеиназ InhVJиз актинии Heteractiscrispa// Биоорган. химия. 2011. Т. 37. С. 310–318.
2.Antuch W., Berndt D. K., Chavez A. M., Delfin J., Wüthrich K. The NMR solution structure of a Kunitz-type proteinase inhibitor from the sea anemone Stichodactyla helianthus // Eur. J. Biochem. 1993. №3(212). P. 675‑684.
3.Bode W., Huber R. Structural basis of the endoproteinase-protein inhibitor interaction // Biochim. Biophys. Acta Protein Struct. Mol. Enzymol. 2000. 1477(1‑2). P. 241–252.
4.Comeau S. R., Kozakov D., Brenke R., Shen Y., Beglov D., Vajda S. ClusPro: Performance in CAPRI rounds 6-11 and the new server // Proteins. 2007. V. 69. №4. P. 781‑785.
5.Delfin J., Martinez I., Antuch W., et. al. Purification, characterization and immobilization of proteinase inhibitors from Stichodactyla helianthus // Toxicon. 1996. V. 34. P. 1367–1376.
6.Farady Ch. J., Craik Ch. S. Mechanisms of Macromolecular Protease Inhibitors // Chem.BioChem. 2010. №11, P. 2341–2346.
7.Guex N., Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling // Electrophoresis. 1997. №15(18). P. 2714‑2723
8.Isaeva M. P., Chausova V. E., Zelepuga E. A., Guzev K. V., Tabakmakher V. M., Monastyrnaya M. M., Kozlovskaya E. P. A new multigene superfamily of Kunitz-type protease inhibitors from sea anemone Heteractis crispa // Peptides. 2012, In Press.
9.Kastritis P. L., Bonvin A. M. Are scoring functions in protein-protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J Proteome Res. 2010 №9(5) 2216‑25.
10.Kiefer F., Arnold K., Künzli M., et. al. The SWISS-MODEL Repository and associated resources // Nucleic Acids Res. 2009. V. 37. P. 387‑392.
11.Krowarsch D., Zakrzewska M., Smalas A. O., Otlewski J. Structure-function relationships in serine protease-bovine pancreatic trypsin inhibitor interaction // Protein Pept. Lett. 2005. V. 12. №5. P. 403‑407.
12.Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. PROCHECK – a program to check the stereochemical quality of protein structures // J. App. Cryst. 1993. № 2(26). P. 283‑291.
13.Peitsch M. C. Protein modeling by E-mail // Nature Biotechnology. 1995. V.13. P. 658‑660.
14.Rawlings N. D., Barrett A. J., Bateman A. MEROPS: the peptidase database // Nucleic Acids Res. 2010, V. 38, D227–D233.
15.Reynolds C. Protor P: a protein-protein interaction analysis server // Bioinformatics. 2009. V. 25, №3. P. 413‑414.
16.Roussel A., Mathieu M., Dobbs A., et. al. Complexation of two proteic insect inhibitors to the active site of chymotrypsin suggests decoupled roles for binding and selectivity // J. Biol. Chem. 2001. V. 276. P. 38893 –38898.
дипломов
Оставить комментарий